Carrier Screening (GxVISION®) Introduction
Carrier screening is genetic testing that determines whether an asymptomatic person has a genetic mutation or abnormalities associated with a particular disorder that may be passed on to children. These are known as recessive conditions.
GxVISION® Carrier Screening Gene Selection Criteria
A carrier panel includes a selected number of recessive genes and conditions. There are different carrier screening panels for you and your provider to choose from. Large panels have more genes and conditions, while we also offer smaller more focused panels. These panels have been developed using guidelines from national expert organizations such as American College of Obstetrics and Gynecology (ACOG), US Department of Health, and American College of Medical Genetics and Genomics (ACMG). To develop their recommendations, they use criteria such as how common a condition is, and how early in life an affected person shows symptoms.
Have a discussion with your physician about which panel would be most appropriate for you. Your doctor may take into consideration your family and medical history. We would encourage you to bring as much information regarding your personal medical and family history as you have available.
If there is a specific syndrome in your family history, records showing the name of the syndrome and the mutation in the family are very helpful to your provider and the laboratory scientists in determining what testing is most appropriate for your history.
GxVISION® Carrier Screening Test Options
Conditions:
Alpha-Thalessemia, Beta Hemoglobinopathies (Beta-Thalassemia and Sickle Cell), Cystic Fibrosis, Duchenne/Becker Muscular Dystrophy, Spinal Muscular Atrophy
Conditions:
Alpha-Thalessemia, Beta Hemoglobinopathies (Beta-Thalassemia and Sickle Cell), Bloom Syndrome, Canavan Disease, Cystic Fibrosis, Deafness, Duchenne/Becker Muscular Dystrophy, Familial Dysautonomia, Fanconi Anaemia, Gaucher Disease, Mucolipidosis IV, Niemann-Pick Disease, Types A and B, Spinal Muscular Atrophy, Tay-Sachs Disease
Genes:
ABCA3, ABCC8, ABCD1, ACADM, ACADVL, ACAT1, AGA, AGXT, AHI1, AIRE, ALDOB, ALPL, ANO10, ARSA, ARX, ASL, ASPA, ATP7B, BBS1, BBS2, BCKDHB, BLM, BTD, CBS, CC2D2A, CCDC88C, CEP290, CFTR, CHRNE, CLCN1, CLRN1, CNGB3, COL7A1, CPT2, CYP11A1, CYP21A2, CYP27A1, CYP27B1, DHCR7, DHDDS, DLD, DMD, DYNC2H1, ERCC2, EVC2, F9, FAH, FANCC, FKRP, FKTN, FMO3, G6PC, G6PD, GAA, GALT, GBA, GBE1, GJB2, GJB6, GLA, GNPTAB, GRIP1, HBA1, HBA2, HBB, HEXA, HPS1, HPS3, IDUA, IKBKAP (ELP1), L1CAM, LRP2, MCCC2, MCOLN1, MCPH1, MID1, MLC1, MMACHC, MMUT (MUT), MVK, NAGA, NEB, NPHS1, NR0B1, OCA2, OTC, PAH, PCDH15, PKHD1, PLP1, PMM2, POLG, PRF1, RARS2, RNASEH2B, RPGR, RS1, SCO2, SLC19A3, SLC26A2, SLC26A4, SLC37A4, SLC6A8, SMN1, SMPD1, TF, TMEM216, TNXB, TYR, USH1b (Myo7a), USH1C, USH2A, XPC (RAD4), FMR1 (optional addition)
Conditions:
Surfactant metabolism dysfunction, pulmonary 3, Familial hyperinsulinism, diabetes mellitus neonatal 3, (X-ALD) X-linked adrenoleukodystrophy, Adrenomyeloneuropathy, Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency, Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, Beta-ketothiolase deficiency (βKT), Aspartylglucosaminuria, Primary hyperoxaluria, type I, Joubert syndrome 3, Autoimmine polyendrocrinopathy syndrome type I, Hereditary fructosuria, Hypophosphaptasia (adult; childhood and infantile), Spinocerebellar ataxia 10, Metachromatic leukodystrophy, Developmental and epileptic encephalopathy 1 (DEE1), Argininosuccinic aciduria (ASA), Canavan disease, Wilson Disease, Bardet-Biedl s yndrome 1, Bardet-Biedl Syndrome 2, retinitis pigmentosa 74, Maple Syrup Urine Disease (MSUD) Type 1B, Bloom syndrome, Biotinidase deficiency (BIOT), Homocystinuria, B6 (HCY), Joubert syndrome 9; Meckel syndrome 6, Congenital hydrocephalus 1, Joubert syndrome 5; Leber congenital amaurosis 10, Cystic fibrosis (CF), Myasthenic syndrome, congenital (4A, slow-channel; and 4B fast-channel), Congenital myotonia, autosomal recessive form, Usher syndrome 3A, Achromatopsia 3, Recessive dystrophic epidermolysis bullosa, Carnitine palmitoyltransferase II deficiency (CPT II), Adrenal insufficiency, congenital, with 46 XY sex reversal (partial or complete), 21-hydroxylase deficiency (CAH), Cererbrotendinous xanthomatosis, Vitamin D-dependent rickets, type 1, Smith-Lemli-Opitz Syndrome, Retinitis pigmentosa, autosomal recessive, congenital disorder of glycosylation type 1, Maple Syrup Urine Disease (MSUD) Type III, Duchenne/Becker Muscular Dystrophy, Short-rib thoracic dysplasia 3 with or without polydactyly, Cerebrooculofacioskeletal syndrome 2; Tricothiodystrophy 1, photosensetive, Chondroectodermal dysplasia, Hemophilia B (HEMB), Tyrosinemia (TYR I), Fanconi Anemia,
Muscular dystrophy-dystroglycanopathy, type A, 5 and type B,5, Fukuyama congenital muscular dystrophy, Trimethylaminuria, Glycogen storage disease, type Ia (GSDIa), Glucose-6-phosphate dehydrogenase deficiency, Pompe disease, glycogen storage disease type II, Galactosemia, type I, Gaucher disease, types I and II, Glycogen storage disease, type IV; GBE1-related disorders, Nonsyndromic hearing Loss, recessive 1a, Nonsyndromic hearing loss, Fabry disease, Mucolipidosis type II alpha/beta, and type III alpha/beta, Fraser syndrome, Alpha thalassemia, Alpha thalassemia, Beta thalassemia, sickle cell disease, Tay-Sachs disease, Hermansky Pudlak syndrome 1, Hermansky Pudlak syndrome 3, Mucopolysaccharidosis Type Ih, Ih/s (Hurler), Familial dysautonomia, Hydrocephalus due to congenital stenosis of aqueduct of Sylvius (HSAS), Donnai-Barrow syndrome, 3-methylcrotonyl-CoA carboxylase 2 deficiency (3MCC), Mucolipidosis type IV, Primary microcephaly 1, recessive, Opitz GBBB syndrome, type 1 (GBBB1), Megalenchepalic leukoencephalopathy with subcortical cysts, Methylmalonic acidemia (cblC) with homocystinuria, Methylmalonic aciduria-methylmalonyl-CoA mutase deficiency, Hyper-IgD syndrome; Mevalonic aciduria, Schindler disease, type 1 and type 3, Nemaline Myopathy 2, Congenital nephrotic syndrome, Adrenal hypoplasia, congenital (AHC), Oculocutaneous albinism, brown and type II, Ornithine transcarbamylase deficiency, Phenylketonuria (PKU), Usher syndrome 1F, deafness autosomal recessive 23, Autosomal recessive polycystic kidney disease, Spastic paraplegia 2, X-linked (SPG2), Congenital disorder of glycosylation type Ia, Mitochondrial DNA depletion syndrome 4A and 4B, Hemophagocytic lymphohistiocytosis, familial, 2, Pontocerebellar hypoplasia type 6, Aicardi Goutieres syndrome 2, Retinitis pigmentosa 3 (RP3; RP); RP X-linked and sinorespiratory infections +/- deafness; Macular degeneration, X-inked atrophic, Retinoschisis 1, X-linked, juvenile (RS1), Mitochondrial complet IV deficiency, nuclear type 2, Basal ganglia disease, biotin-responsive, Epiphyseal dysplasia, mulitple 4; Achondrogenesis 1b, Pendred Syndrome, Glycogen storage disease 1b and 1c, Cerebral creatine deficiency syndrome 1 (CCDS1), Spinal Muscular Atrophy, Niemann-Pick disease (Type A & B), Atransferrinemia, “Joubert syndrome 2,, Meckel syndrome 2”, Ehlers-Danlos like syndrome due to tenascin-X deficiency, Oculocutaneous albinism type 1A and 1B, Usher syndrome, nonsyndromic hearing loss, Usher Syndrome, Usher syndrome, type 2A, Xeroderma pigmentosum, Fragile X Syndrome (FXS) (optional addition)
How are Recessive Conditions Passed Along in Families?
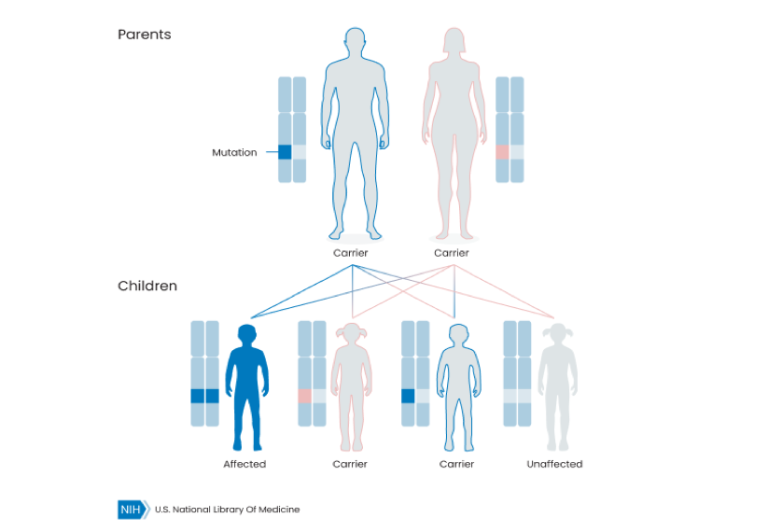
Most recessive conditions require that both biological parents carry a mutation. Those are known as autosomal recessive. If a couple carries an autosomal recessive condition, there is a 25% chance that their child will be affected by the condition.
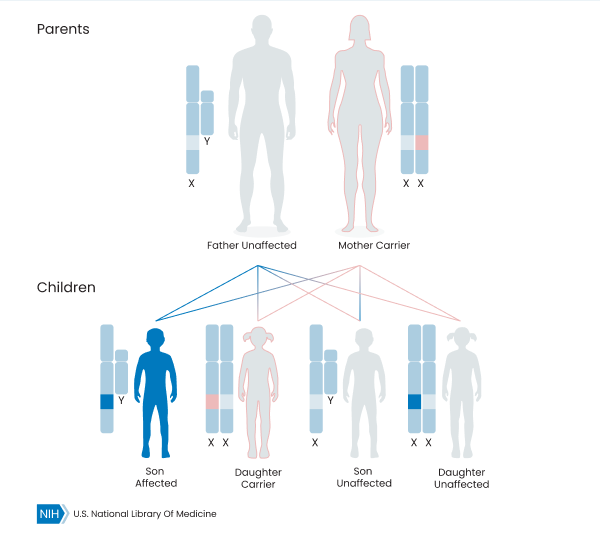
For other recessive conditions, a person’s biological gender influences whether they have the disorder or not. These are called X-linked conditions. Women who inherit these mutations are carriers, while men who inherit the genetic mutation have the condition, even if their male parent does not carry the mutation. Occasionally, carriers of recessive conditions can have some symptoms related to the mutation, but these are usually more mild than the symptoms seen in the disorder.
See below an illustration of some of the inheritance patterns.
Autosomal Recessive
X-Linked Recessive
What Sort of Results Should I Expect?
We report Positives, Negatives and Variants of Unknown Significance (VUS). Most people carry at least one recessive condition. Being a carrier does not necessarily mean that there will be medical issues for you or your children/ future children.
- Positive: You were shown to be a carrier for a genetic condition.
- Negative: You are likely not a carrier for the tested conditions.
Rarely, a Variant of Uncertain Significance (VUS) will be reported for carrier screening*. VUS: A difference was found in your DNA that may or may not cause you to be a carrier. A VUS is reported when it is linked to an increased carrier risk and/or there is actionable clinical follow-up.
*All VUSs are available upon request.

Education Center
Stay informed with our patient resources. Visit our Education Center to learn more.
FAQ’s Billing & Insurance FAQs
Every patient should have access to highly accurate and affordable genetic testing. Otogenetics is committed to work with each patient to reach payment solutions. These may include:
- Working with your insurance provider to verify coverage.
- Flexible payment options billed over several months.
- Financial assistance for patients who qualify.
We encourage patients to call us for all billing questions, and to discuss payment solutions.
Please call us toll free at 1-855-686-4363, option 1.
Otogenetics is committed to working with patients to ensure that everyone has access to testing without financial burden.
- Medicaid patients will NOT have patient out of pocket responsibility.
- We treat all out-of-network claims as if they are in-network.
- We will work with all insurers (primary and secondary policies) to maximize your qualified coverage.
Otogenetics testing service is covered by many insurance plans, the exact amount you owe may vary based on your individual plan and financial situation.
Here is our commitment to you regarding costs:
- We will reach out to you if we estimate your cost could be more than $50 (the average out of pocket Otogenetics patients pay).
- We will discuss the estimate with you, and review your affordability.
You may receive information from your insurer, known as an Explanation of Benefits (EOB), regarding the test you received from Otogenetics.
An EOB from your insurer is NOT A BILL.
A bill for the test(s) provided by Otogenetics comes ONLY from Otogenetics.
If there is any patient out of pocket cost after Otogenetics has exhausted all options for reimbursement coverage and financial assistance, Otogenetics will reach out to you, the patient.
Next Steps

To get started, talk to your healthcare provider about requesting a carrier screen.

Your healthcare provider will take a blood draw or buccal swab and send it to our lab.

Otogenetics will analyze your DNA sample using our state-of-the-art technologies.

Once your screen is complete, your healthcare provider or a genetic counselor from Otogenetics may contact you to explain your results and answer any questions you have.